Реферат: Анализ следов веществ
Реферат: Анализ следов веществ
Анализ следов веществ
Термины
макро и микро
применяются в химическом анализе как для выражения размера
пробы, так и для выражения относительного количества компонента, подлежащего
определению. На заре развития аналитической химии использовались относительно
большие пробы, поэтому не требовалось никакого особого обозначения для
определения масштаба аналитических операций. По мере возрастания требований
науки и техники и прогресса аналитических методов стали возможны химические
определения на пробах в миллиграммовых количествах вместо десятков граммов; с
тех пор вошел в обиход термин микроанализ в
отличие от старого, называемого в настоящее время макроаншизом.
Хотя абсолютные количества веществ, с которыми приходится
оперировать в макро- и микроанализе, различаются в 10 и 100 раз, относительные
количества определяемых компонентов остаются темп же, т.е. больше 0,01 пли даже
0,1. Химические определения ниже этого предела выполнялись редко как из-за
отсутствия потребности, так и вследствие трудностей анализа такого количества
вещества существовавшими методами. Известно, что затем был введен термин следы
для обозначения ничтожного количества вещества,
присутствующего в пробе, но точно не определяемого. Положение в настоящее время
значительно изменилось. Теперь потребности науки и техники в определении
компонентов, составляющих лишь малую долю анализируемого образца, сильно
возросли, и желательно следы вещества определять более точно. Как с логической,
так и с исторической точки зрения имеется веское основание для установления
верхнего предела содержания следов вещества, или микрокомпонента, равного 0,01%.
В связи с этим представляет интерес утверждение, сделанное в отношении анализа
горных пород: «Относительно термина «следы» можно сказать, что под ним
подразумевают такую концентрацию вещества, которая находится ниже предела
количественного определения его в образце, взятом для анализа. Для анализов,
претендующих на полноту и точность, в общем случае следует указать, что
предполагаемое содержание интересующего компонента меньше 0,02 или даже 0,01%».
Значение
концентрации 0,01% приблизительно соответствует нижней границе чувствительности
обычно применяемых весового и объемного методов анализа, поэтому мы можем
принять его как ориентировочно указывающее верхний предел содержания следов
вещества. Нет никакой необходимости для установления жесткой концентрационной
границы для микрокомпонента. Иногда удобно рассматривать как следы примесь,
которая содержится в количестве нескольких сотых долей процента. Так, например,
в силикатных породах содержание меди обычно составляет 0,001–0,05%, однако
можно говорить о меди как о следах примеси в этом материале.
Чтобы
уменьшить резкость перехода от микро- к макрокомпонентам, удобно подразделить
последние на главные и второстепенные. Область второстепенного макрокомпонента
лежит в пределах от 0,01 до 1%. Она имеет, по крайней мере, у своей нижней
границы, несомненное сходство с областью микрокомпонента, и для определения
второстепенного макрокомпонента часто применяют методы, используемые при
анализе следов веществ.
Содержание
главного или второстепенного компонента в образце обычно выражают в весовых
процентах. Те же единицы часто используют для выражения содержания следов
вещества, но удобнее давать их содержание в частях на миллион, особенно для
концентраций ниже 0,001%.
Существенная
особенность анализа следов веществ состоит не просто в определении малого
количества вещества, а в его обнаружении в присутствии подавляющего количества
других веществ, которые могут серьезно повлиять на реакции определения
микрокомпонентов. Анализ следов веществ имеет характерные черты как макро-, так
и микроанализа. Величина пробы, а иногда и предварительные стадии проведения
анализа следов вещества подобны тем, которые используются при макроанализе.
Иногда для осуществления анализа следов вещества требуется больший исходный
образец, чем используется при макроанализе. В редких – случаях употреблялись
образцы, весящие несколько сот килограммов. Конечная стадия анализа при
определении следов веществ может иметь более микроскопический характер, чем в
обычном микроанализе, где 1 у является
пределом желаемой точности определения. Многие колориметрические методы
позволяют определять 1 у или
меньшее количество вещества с точностью 5–1096; то же самое относится к
спектральному методу анализа. В радиоактивационном анализе приходится иметь
дело с еще меньшими количествами вещества.
Таблица 1. Распространение некоторых
редких элементов в вулканических породах
| Элеыент |
Содержание,
ч. на ылн. |
Элеыент |
Содержание,
ч. на млн. |
| Ag |
0,1 |
Nb |
20 |
| As |
2 |
Ni |
80 |
| Au |
0,005 |
P |
800 |
| В |
3 |
Pb |
15 |
| Ва |
250 |
Pd |
0,01 |
| Be |
2 |
Pt |
0,005 |
| Bi- |
0,2 |
Rb |
310 |
| Cd |
0,15 |
Re |
0,001 |
| Се |
46 |
S |
520 |
| CI |
200 |
Sb |
0,1 |
| Со |
23 |
Sc |
5 |
| Cr |
200 |
Se |
0,09 |
| Cs |
7 |
Sn |
2 |
| Си |
70 |
Sr |
450 |
| F |
300 |
Та |
15
(?) |
| Ca |
15 |
Те |
0,002
(?) |
| Ge |
1.5 |
Th |
11,5 |
| Hf |
4,5 |
Ti |
4400 |
| Hg |
0,5 |
Tl |
0,3 |
| J |
0,3 |
U |
4 |
| In |
0,1 |
V |
150 |
| La |
18 |
W |
1 |
| Li |
65 |
Y |
21 |
| Mn |
1000 |
Zn |
80 |
| Mo |
1 |
Zr |
220 |
Рассмотрение
распределения следов элементов в природе не входит в план настоящей книги,
однако небезынтересно привести таблицу распространения редких элементов в
вулканических породах. Некоторые из указанных в ней значений являются
приближенными. Для большинства элементов среднее содержание в земной коре такое
же, как и в вулканических породах. Многие из так называемых редких элементов
встречаются в природе столь же часто, как и элементы, которые обычно считают
распространенными. Германий имеется примерно в таком же количестве, как и
мышьяк, галлий – как свинец, церий – как цинк; скандием природа богаче, чем
ртутью или висмутом, и т.д. Редкие элементы тем или
иным
путем переходят из вулканических пород в растения или организмы животных, и
многие из них с течением времени концентрируются в живой материи. Важная роль,
которую играют в биологических процессах следы элементов, теперь хорошо
известна, и их определение представляет практический и научный интерес.
Методы анализа следов веществ
При
определении следов элементов использовались и используются методы, аналогичные
тем, которые применяют в общем химическом анализе. Следы веществ определяли
даже весовыми методами. Например, весовым методом был определен галлий в образце
алюминия весом 50 г. с
содержанием Ga
менее 0,001%. Для анализа следов веществ в верхней концентрационной области
иногда можно эффективно применять объемные методы, особенно в тех случаях,
когда конечная точка титрования находится электрометрически. При определении
следов тяжелых металлов иногда применяют экстрактивное титрование раствором
дитизона в органическом растворителе. Однако, когда возникает необходимость
определения весьма малых количеств веществ, обычно применяют ие весовой или
объемный, а другие методы анализа. Наиболее важные из них, распространенные в
настоящее время, перечислены ниже.
1. Колориметрический,
спектрофотометрический и связанные с ними методы.
2. Метод
оптической спектрографии.
3. Метод
рентгеноспектроскопии, основанный на поглощении и испускании рентгеновских
лучей.
4. Радиоактивационный
метод.
5. Масс-спектрометрический
метод.
6. Полярографический
и другие электрохимические методы.
7. Метод
катализа и индукции.
8. Другие
методы.
Некоторые
из этих методов обладают не только высокой чувствительностью, но и пригодны для
определения. следов большинства элементов. Другие методы не имеют достаточной
чувствительности, и ценность их применения в области концентраций несколько
частей на миллион ограничена. Часть методов пока недоступна для обычных
аналитических лабораторий.
Радиоактивационный
анализ. До тех пор пока определение элементов путем
измерения радиоактивности ограничивалось естественными радиоактивными
элементами, оно представляло незначительный интерес. Положение существенно
изменилось в настоящее время, когда можно получать радиоизотопы большинства
элементов путем нейтронного облучения последних в ядерном реакторе. Метод
нейтронной активации применяется для определения следов элементов во
всевозрастающем масштабе. Нижний предел радиоактивационного определения
составляет 0,01–0,0001 ч. на млн. При
выполнении активационного анализа взвешенный образец облучается нейтронами в
ядерном реакторе в течение определенного периода времени. В процессе облучения
образуется один или несколько радиоизотопов исследуемого элемента.
Радиоактивность А за время t после
начала нейтронной бомбардировки вычисляется по уравнению
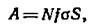
где
Л – число
распадов в 1 сек; N –
число атомов мишенных изотопов; f – нейтронный поток на 1 снг
в сек; а –
поперечное сечение активации, смг1атом; S –
коэффициент насыщения, равный 1-е-', или отношение радиоактивности,
наведенной за время t,
к радиоактивности, возникшей за бесконечно большое время.
Если
скорость накопления радиоактивности Nfo постоянна во время облучения, то приведенное
выше уравнение можно записать как
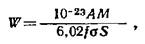
где
М – химический атомный вес
элемента, W – навеска мишени, г.
Поскольку
нейтронный поток трудно измерить или проконтролировать, одновременно с самим
образцом облучают стандарт, содержащий известное весовое количество элемента.
Тогда отношение количеств анализируемого элемента в стандарте и образце
характеризуется отношением их активностей. Обычно активационное определение
элемента осуществить не так просто, как это может показаться, ибо другие
составные части образца могут также сильно активироваться в процессе облучения,
и тогда анализируемый элемент приходится химически отделять от других
радиоактивных изотопов. Поэтому как к стандарту, так и к образцу после
облучения перед операцией химического отделения необходимо добавить известное
количество неактивного элемента. В том случае, если извлечение не окажется
количественным, следует ввести поправку.
По
литературным данным, для потока нейтронов из ядерного реактора плотностью 5–10 нейтрон
1 см-сек чувствительность актнвацион-ного метода
для различных элементов выражается следующими значениями: V – 0,003 у; Мп – 0,001
у; Sr –
0,6 у; Со – 0,04 у;
Ga –
0,01 у; Cd –
0,10 у. Как утверждают,
точность определений превышает 10%.
Ряд
элементов – Li,
Be, Mg, Al,
Nb, Ti, Rh – не удается определить с достаточной чувствительностью
методом активации нейтронами.
Масс-спектромстрия.
Применение метода изотопного разбавления в соединении с масс-спектрометрией
позволяет производить определение ряда элементов в твердых образцах с
концентрацией до 10~. После того как образец введен в раствор, добавляется
известное количество изотопного индикатора определяемого элемента. Затем
элемент отделяют химически; изменение в изотопном составе, обусловленное
индикаторным разбавлением, определяют масс-спектрометрнчески и таким образом
находят первоначальное количество элемента. Таким способом успешно определяли
уран в каменных метеоритах с концентрацией до 0,01 ч. на млн.
Около
7096 всех элементов имеет несколько стабильных изотопов и могут, по крайней
мере в принципе, определяться методом изотопного разбавления. Современные
масс-спектрометры дают возможность анализировать металлы и тугоплавкие вещества
при температурах вплоть до 2500°; таким образом, теперь приготовление
соединений с высокой упругостью пара при низкой температуре не так важно, как
раньше.
Полярография
в. Большинство аналитиков
хорошо знакомо с принципами полярографического метода, и здесь необходимо лишь
сказать, что по числу элементов, которые можно определять таким путем, по
чувствительности и точности определений он конкурирует с колориметрическим и
спектрометрическим методами, иногда даже превосходя их.
Катализ
и индукция. Некоторые элементы можно определять путем
ускорения ими окислительно-восстановительных реакций, в обычных условиях
протекающих очень медленно. Например, осмий, рутений и иод сильно катализируют
медленную реакцию
As+2Ce -» As+2Ce.
Катализирующие
элементы с более высоким состоянием окисления быстро восстанавливаются мышьяком
до состояний, которые вновь быстро реокисляются церием. Скорость катализируемой
реакции пропорциональна концентрации катализатора и обычно очень легко
определяется фотометрически путем измерения светопоглощения желтой окраски
раствора в зависимости от времени. Осмий и иод можно обнаружить в растворах,
разбавленных до концентраций 0,001 и 0,01 ч. на млн.'
Примером
реакции индуцирования, которую можно применить для определения следов вещества,
служит восстановление хрома четырехвалентным теллуром до более низкого
состояния окисления, которое, по-видимому, соответствует четырехвалентному
хрому:
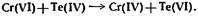
При
избытке хрома эту реакцию можно использовать для определения теллура
титрованием, осуществляя взаимодействие Cr с ионами фенантролина железа в растворе
соответствующей кислотности:
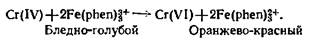
Количество
образовавшегося фенантролина железа, которое можно найти спектрофотометрическим
измерением, соответствует эквивалентному количеству теллура. Этот косвенный
метод определения теллура более чувствителен, чем любой прямой
колориметрический метод, доступный для данного элемента.
Если
в избытке находится теллур, то Сг, образовавшийся по реакции, вновь
восстанавливается теллуром в хром, который восстанавливает фенантролин железа, и
т.д. Эта реакция сочетает индукцию и катализ. Она позволяет обнаружить
чрезвычайно малые количества хрома. Ланг приводит предельное значение
концентрации при определении хрома, равное 1:2–10. Циклические реакции в конце
концов завершаются благодаря побочным реакциям, например реакции между Те и Сг с
образованием Cr.
Каталитические
индуцированные реакции не имеют большого практического значения, если нет
эффективного средства отделения анализируемого вещества.
Другие
методы. Микроскопические методы, основанные на измерении
диаметра королька золота, получаемого в результате купеляции, или шаровидных
частиц ртути, нашли ограниченное применение.
Нехимический
метод определения следов некоторых элементов, являющихся питательными
веществами для бактерий, основан на избирательном росте микроорганизмов по
отношению к ним.
Роль колориметрии в анализе следов веществ
В
настоящее время основная масса анализов по определению следов веществ
выполняется при помощи эмиссионной спектрографии и колориметрии.
Спектрографический метод применим для определения любого элемента, однако с «чувствительностью,
изменяющейся в широких пределах. Для некоторых элементов нет удовлетворительных
колориметрических методов определения, для других эти методы недостаточно
чувствительны, чтобы их использовать в анализе следов веществ.
Колориметрическому определению лучше всего поддаются тяжелые металлы. Как
правило, колориметрическое определение следов элементов требует проведения
многочисленных операций разделения. В этом требовании заключается как слабая,
так и сильная стороны метода. С одной стороны, не всегда есть эффективные
методы разделения. В процессе разделения могут происходить незначительные
потери определяемого компонента и не полностью удаляться элементы, мешающие
определению. Процедура отделения следов элемента может оказаться довольно
трудной. С другой стороны, если возможно осуществить удовлетворительное
отделение – а это в действительности скорее правило, чем исключение, – влияние
посторонних элементов устраняется, и колориметрический метод становится
абсолютным. Этого часто нельзя сказать в отношении обычных спектрографических
анализов, в ходе которых не делается никаких химических разделений, и точность
результата может сильно зависеть от состава образца и от точности стандарта.
Кроме того, точность колориметрического определения может превысить точность
спектрографического определения; проще измерить оптическую плотность раствора,
чем плотность линии на фотографической пластинке. Таким образом, выигрыш во
времени, который дает спектрографический метод, может быть сведен на нет
меньшей надежностью и точностью определений. Спектрографический метод анализа
наиболее предпочтителен в тех случаях, когда приходится производить много
определений одного или нескольких элементов на образцах примерно постоянного
макроскопического состава. При небольшом числе измерений значительная работа,
которую нужно проделать при калибровке прибора, вряд ли стоит затраченного
времени. В этом случае более удобными могут оказаться колориметрические методы.
По концентрационной чувствительности спектральный и колориметрический методы
часто имеют меньшие различия, чем обычно считают. В спектрографическом методе
анализа обычно применяют образец микроскопического размера, а это ведет к
увеличению минимального процентного содержания элемента, которое можно
определить, даже если абсолютная чувствительность метода высока. При
колориметрическом определении величина анализируемого образца может быть в 100
раз больше, чем при простом спектрографическом определении, и поэтому более
высокая относительная или концентрационная чувствительность колориметрического
метода может уравновесить более высокую абсолютную чувствительность
спектрографического метода. Если имеющееся в распоряжении количество
анализируемого вещества ограничено, относительная чувствительность спектрального
метода может, конечно, превзойти чувствительность колориметрического метода, но
положение часто становится обратным, если доступны 1–2 г. вещества.
Интересно провести некоторые сравнения спектрального и колориметрического
методов по их чувствительности. Значения средней абсолютной чувствительности
спектрального метода при различных способах возбуждения приведены ниже.
| Способ
возбуждения |
Количество
металла на электроде, мг |
| Дуга
постоянного тока |
10–5-Ю-4 |
| Катодный
слон дуги постоянного тока |
10-в_ш-5 |
| Высоковольтная
дуга переменного тока |
Ю-» – ю-5 |
| Конденсированная
искра постоянного тока |
1
о-» – ю-* |
Практическая
чувствительность некоторых цветных реакций соответствует 10
мг или меньше.
Метод
дуговой спектрографии обычно применяют для определения следов элементов в
силикатах. Чувствительность спектрального метода, выраженная в частях на
миллион, при определении различных металлов указана ниже: Ag 1; Be
10, 1, 4; Cd
300; Со 2, 10; Сг 1, 1, 0,2; Ga
5, 10, 5; Ge
15; Mo 1, 5; Ni 2, 5; Pb 30, 10, 10; Zn 350, 100. Каждый из этих металлов можно
определить колориметрически или флуо-риметрически с чувствительностью, равной 1 ч.
на млн.
Посредством
специальной методики, заключающейся во фракционной перегонке, некоторые
относительно легко летучие металлы, такие, как цинк, кадмий, ртуть, индий,
таллий, германий, мышьяк и висмут, можно обнаружить спектрографически в
количестве порядка 0,01 ч. на мл при анализе образца силиката весом 1–3 г.
Вообще чувствительность спектрографического анализа можно
повысить, если элемент, подлежащий определению, предварительно отделить, так
как абсолютная чувствительность спектрографического метода обычно выше по
сравнению с колориметрическим методом. В прошлом и в значительной степени в
настоящее время аналитики-спектрографисты в целом довольно редко прибегали
к
этому способу, хотя время от времени он все же применяется. Благодаря высокой
разрешающей способности спектрографического метода часто следует предпринимать
лишь частичное разделение в форме химического обогащения определяемых
компонентов. Для разделения применяют осаждение, соосаждение, экстракцию
несмешивающимися растворителями, а также другие методики.
Общий
предел особо чувствительных колориметрических методов составляет около 0,1 ч.
на млн. при анализе твердого образца весом 1 г. Достижение
этого предела с образцами сложного состава предполагает доступность хороших
методов разделения. При анализе образцов большего веса предел измерений может
быть снижен. Селен в почвах можно определить колориметрически с чувствительностью
до 0,0001 ч. на млн. Sa Предел измерения
содержания некоторых тяжелых металлов в водных растворах, пробы большого объема
которых легко обрабатываются, составляет 10 ч. на млн. При анализе
биологических материалов возможная концентрация обнаружения ряда элементов
менее 0,1 ч. на млн.
Для
большинства элементов радиоактивационный метод анализа значительно превосходит
по чувствительности спектрографический и колориметрический методы. Надежность
определения по сравнению с колориметрией также может быть выше, потому что
достоверность выделенного радиоэлемента можно проконтролировать путем измерения
его периода полураспада, вносится поправка на потери при выделении
радиоэлементов, а возможность загрязнения посторонними примесями ограничена,
так как не требуется какой-либо химической обработки препарата до активации. Но
в то же время работа с радиоактивными веществами требует соблюдения мер
предосторожности, химическая обработка радиоактивного образца часто трудна и
длительна, в ряде случаев отсутствует избирательность. Радиоактивационный
анализ особенно ценен для определения элементов, содержащихся в препарате в
количестве менее X,
l ч.
на млн. В общем этот метод более приемлем для решения специальных проблем, чем
для выполнения обычных анализов.
Широкое
применение колориметрического метода для определения следов и незначительных
количеств веществ объясняется рядом причин: умеренными требованиями к
аппаратуре, возможностью использования метода аналитиком средней квалификации,
высокой чувствительностью метода, отвечающей современным требованиям, и
точностью, не менее высокой, чем точность любого другого метода в этой области.
В
табл. 2 и 3 приведено несколько сравнительных оценок по определению следов
веществ в материалах биологического происхождения различными методами. Можно
установить хорошее соответствие полученных данных с результатами
колориметрических измерений.
Меры предосторожности при определении следов веществ
Попадание
посторонних веществ во время приготовления пробы и в ходе самого анализа может
иметь более серьезные последствия при анализе следов, чем в каком-либо другом
типе анализа; на этот источник ошибок следует обратить особое внимание.
Необходимо
тщательно соблюдать меры предосторожности для предохранения пробы от
существенного загрязнения в результате случайного попадания металла из общих
лабораторных предметов: железа из штативов, никеля из тигельных щипцов, меди и
цинка из горелки, цинка из резины и т.д. При просеивании нужно использовать
шелковые сита вместо металлических. Следует иметь в виду возможное попадание
некоторых легирующих элементов стали, когда для измельчения твердых материалов,
таких, как силикаты, пользуются ступкой Плетнера из закаленной стали.
Количество посторонних металлов, которые могут таким путем попадать в препарат
в обычных условиях измельчения в ступке, указано в табл. 4. Иногда встречаются
и некоторые непредвиденные элементы. Так, например, в стали ступки Плетнера
были найдены ниобий и тантал».
Описаны
плитки и дробитель из высококачественной алюминиевой керамики для измельчения
горных пород. Как установлено, единственными элементами, которые попали в
порошок при измельчении куска кварца до 100 меш, были титан и магний; алюминия
найдено не было.
Серьезное
загрязнение растительных материалов может происходить при их механическом,
измельчении. Так, установлено, что при размалывании образцов на молотковых
мельницах и мельницах системы Wiley
вносятся железо и медь. Применение вибромельницы с кремневыми шарами приводило
к загрязнению железом, медью, цинком, кобальтом и натрием; при использовании
фарфоровых или муллитовых шаров, помимо этих элементов, вносятся также кальций,
сера и фосфор. Измельчение вручную пестиком в фарфоровых ступках не приводило к
заметному загрязнению пробы железом, медью, цинком, бором, кобальтом, марганцем,
молибде-hqm, кальцием, натрием, магнием, фосфором,
серой или калием. Сконструирована мельница с нейлоновыми роликами, которая
должна обеспечить приготовление растительных образцов без заметного загрязнения
следами элементов.
Малые
количества некоторых тяжелых металлов могут попадать в препарат из стеклянной и
платиновой посуды, применяемой для анализа. Так, стекло пирекс может давать
следы мышьяка, цинка, свинца, а возможно, и других тяжелых металлов. Вообще
стекло пирекс и другое боро-силикатиое стекло не применяются при определении
малых количеств бора; для этой цели подходит стекло корнинг №728. Платиновая
посуда обычно содержит железо, и некоторое количество его почти наверняка будет
попадать в кислые растворы при контакте с платиной. Глазурь фарфора может
содержать такие тяжелые металлы, как свинец. Посуда из плавленого кварца часто
представляет большую ценность для анализа следов веществ, так как она вообще не
содержит тяжелых металлов и химически стойка к большинству кислот. Иногда
находит применение стекло викор. Оно не содержит щелочных металлов и кальция,
но в нем имеются R203, В и As.
Анализ
следов веществ не следует выполнять в стеклянной посуде, которая ранее
использовалась для макроанализов. Так, при определении следов молибдена
совершенно недопустимо применять стеклянный стакан, в котором ранее
производилось осаждение фосфора в вндефосфоромолибдата аммония. Стеклянная
посуда, обработанная хромовой смесью, прочно удерживает следы хрома даже после
тщательной промывки. Фильтровальная бумага всегда содержит небольшие количества
металлов. Так, в беззольной фильтровальной бумаге были найдены следующие
элементы:
Al, Ba, Ca, Cr, Cu, Fe, Ge, K. Mg, Mn, Na, Ni, Pb, Sb, Si, Ti. V,
Zn, Zr и редкие земли. Некоторые из этих
элементов содержатся в ничтожных, другие – в заметных количествах. Например, в
золе беззольных фильтров было найдено около 1% РЬО, 0,3% ZnO и 0,3% Sn02; всегда присутствуют большие
количества кальция, магния и других элементов.
Некоторые
летучие вещества могут попадать в анализируемый образец из атмосферы
лабораторного помещения; они поглощаются жидкостями и твердыми веществами. Вряд
ли следует упоминать о мерах предосторожности, которые нужно предпринять, чтобы
исключить попадание пыли на различных стадиях анализа. В атмосферной пыли могут
находиться в заметных количествах элементы, считающиеся нераспространенными.
Так, в пыли было найдено до 300 ч. на млн. мышьяка. Отмечено загрязнение
образцов цинком и магнием из^ пудры и другой косметики.
Возможна
также потеря следов определяемого элемента вследствие взаимодействия его с
материалом стенок сосуда. Потери за счет адсорбции стеклом сосудов рассмотрены
в другом разделе. Фильтровальная бумага может адсорбировать из растворов такие
металлы, как свинец и медь, особенно если растворы нейтральны или имеют
слабокислый характер; вообще предпочтительны фильтры из неорганических веществ.
К нерастворимому материалу на любой стадии анализа следует относиться с особым
вниманием. Остаток может удерживать тяжелые металлы, причем иногда в больших
количествах. Примером служит удерживание железа и алюминия и, в значительно
меньшей степени, марганца двуокисью кремния, образующейся при сухом озолении
органических материалов, содержащих кремнезем. Нерастворимую двуокись кремния
нужно подвергнуть соответствующей обработке, чтобы извлечь любой из
содержащихся в ней определяемых элементов.
Таблица 5. Содержание металлов в
воде при различных способах дистилляции
| Способ
днстнлляцнн |
Содержание
металла, у/л |
|
|
Си |
Zn |
Мп |
Fe |
Mo |
| В
луженом медном пере- |
|
|
|
|
|
| гонном
аппарате…. |
10 |
2 |
1 |
2 |
2 |
| Вода
из медного перегон- |
|
|
|
|
|
| ного
аппарата после |
|
|
|
|
|
| однократной
дистилля- |
|
|
|
|
|
| ции
в аппарате нз стек- |
|
|
|
|
|
| ла
пирекс после двух |
1 |
0,12 |
0,2 |
0,1 |
0,002 |
| кратной
дистилляции в |
|
|
|
|
|
| аппарате
из стекла пи- |
|
|
|
|
|
|
|
0,5 |
0,04 |
0,1 |
0,02 |
0,001 |
| после
трехкратной ди- |
|
|
|
|
|
| стилляции
в аппарате |
|
|
|
|
|
| из
стекла пирекс… |
0,4 |
0,04 |
0,1 |
0,02 |
0,001 |
Реагенты
При
определении обычных тяжелых металлов, таких, как железо, медь, цинк и свинец,
может возникнуть необходимость в предварительной очистке применяемых реагентов.
Особое внимание следует обращать на чистоту дистиллированной воды. Обычная
дистиллированная лабораторная вода часто содержит относительно большие
количества некоторых металлов, и поэтому ее нельзя применять для анализа следов
металлов i.
Достаточно чистую для большинства целей воду можно получить простой повторной
дистилляцией в перегонном аппарате, изготовленном целиком из стекла пирекс. В
воде двухкратной дистилляции содержание свинца не более 1 у
1 л. Ее можно хранить в сосудах из стекла пирекс или
другого устойчивого стекла. Для удаления следов металлов из дистиллированной
воды можно использовать ионообменные смолы. Присутствие в воде органических
веществ и других примесей, обладающих свойствами восстановителя, недопустимо
при некоторых анализах, например при определении хрома дифенилкарбазидом. В
дистиллированной воде возможно также накопление хлора, которое может вызвать
трудности при некоторых определениях следов веществ.
Обычные
кислоты, как правило, содержат очень мало тяжелых металлов, однако в ряде
случаев требуется их очищать, когда кислоты применяют в больших количествах.
Обычно дистилляцией кислоты в перегонном аппарате из стекла пирекс удается
получить продукт, достаточно чистый почти для всех целей. Хлорную кислоту в
случае необходимости можно вторично перегнать при пониженном давлении.
Плавиковую кислоту можно очистить от свинца, соосаждая фторид свинца с фторидом
стронция. Чистую гидроокись аммония проще всего получить насыщением
бидистиллята аммиаком в холодной ванне. Гидроокись аммония рекомендуется
хранить в полиэтиленовой посуде или в покрытых церезином склянках. Очень чистую
гидроокись аммония получают изотермической перегонкой. Для этого в пустой
эксикатор помещают два стакана: один с концентрированным аммиаком, другой с чистой
водой и оставляют примерно на день. Аналогично можно приготовить чистую соляную
кислоту.
Растворимые
твердые вещества очищают перекристаллизацией для более или менее полного
удаления всех посторонних веществ или же химической обработкой для удаления определенных
загрязнений. Метод химической обработки применяется часто. К раствору можно
добавить подходящий реагент, который образует малорастворимое соединение с
веществом, подлежащим удалению; целесообразно применять по возможности такой
носитель, который образует смешанные кристаллы с осаждаемым соединением.
Применяют также экстракционные методы очистки. Например, ряд тяжелых металлов
можно извлекать из нейтральной или слабощелочной среды с помощью дитизона.
Холостые опыты
Правильно
поставленный холостой опыт дает представление о количестве определяемого
вещества, вносимого сосудом и реагентами. Цель очистки реагентов заключается в
уменьшении содержания анализируемого компонента до такой степени, чтобы оно
было мало по сравнению с содержанием его в пробе; по возможности посуда,
употребляемая в анализе, должна быть из такого материала, который не будет
отдавать в раствор заметного количества определяемого вещества.
Холостой
опыт необходимо провести по всем соответствующим стадиям анализа. Большая
величина, получаемая в холостом опыте, естественно, нежелательна, так как она
снижает точность определения компонента в пробе. Если известно, что никакого
заметного количества определяемого вещества с химической посудой не вносится,
целесообразно для увеличения точности провести холостой опыт с большим
количеством реагентов. Холостой опыт, в котором определяемый элемент не найден,
безусловно, не является доказательством того, что его на самом деле нет в
растворе. Такой результат при анализе следов может вызвать подозрение. Он может
означать, что посторонние вещества, присутствующие в реактиве, соединяются с
исследуемым компонентом, не допуская его взаимодействия с колориметрическим
реагентом. Например, следы сульфида при определении различных тяжелых металлов
дитизоном могут привести к заниженным результатам, образуя соединения, инертные
к колориметрическому реагенту. Присутствие таких мешающих веществ можно
обнаружить, подвергнув стандартные растворы той же обработке, что и
анализируемый образец.
Стандартные
растворы
Разбавленные
растворы, содержащие ионы металлов, могут значительно уменьшать свою
концентрацию при стоянии в результате адсорбции на стеклянных стенках сосуда
или катионного обмена с ними. Потерн металла наиболее высоки в нейтральной или
слабощелочной среде, но они могут происходить также в слабокислых растворах,
хотя и в меньшей степени. Чем сильнее разбавлен раствор, тем большим будет
относительное уменьшение концентрации. Так, найдено г, что в
растворах Mo,
V, Ti и Ni с концентрацией 0,001% содержание металла
после 75 дней хранения раствора в хорошо промытом сосуде из венского стекла
составило от 0,2 до 0,4 от первоначального; в растворах Au, Pt,
Pd и Ru с концентрацией 0,001% содержание металла
через 230 дней хранения в посуде из того же стекла уменьшалось до 0,1–0,3 от
первоначальной концентрации. В сосудах из кварцевого стекла наблюдалось лишь
незначительное уменьшение концентрации растворов, за исключением раствора
палладия, концентрация которого после стояния в течение 230 дней упала до 0,3
первоначальной величины. Согласно данным, полученным автором, в кислых
растворах, которые хранят в сосудах из стекла пирекс, для большинства металлов
потери гораздо меньше указанных; однако этот источник ошибок не следует
недооценивать. Рекомендуется готовить стандартный раствор в два приема. Готовят
раствор средней концентрации в 0,1–1 н. кислоте. Этот раствор должен быть
стабильным в течение длительного времени, особенно если содержится в сосудах из
стекла пирекс; из этого раствора разведением можно получить более разбавленные
стандартные растворы. Их готовят слабокислыми и хранят в сосудах из стекла
пирекс. В растворах не должно происходить каких-либо заметных изменений
концентрации в течение 1–2 недель. Даже очень разбавленные растворы свинца и
ионов уранила устойчивы при хранении в колбах из боросиликатного стекла или
полиэтилена в течение нескольких месяцев, если их кислотность достаточна для
предотвращения гидролиза. Особенно важно подкнсление стандартных растворов
металлов высокой валентности. Для сохранения некоторых металлов, таких, как
цирконий, иногда необходима кислотность до 1
или 2 н.
Рост
плесени в растворах органических кислот может привести к потере металлов из
раствора, как это показано на примере стронция.
Растворы
металлов в органических растворителях никогда не хранят в полиэтиленовых
бутылях. Стандартные растворы можно готовить из чистых металлов или чистых
солей определенного состава. Если имеются соли хорошего качества, их, как правило,
можно применять для колориметрии без дополнительной очистки. По возможности
применяют безводные соли. Чтобы удалить большую часть связанной воды, соли
следует измельчить до порошкообразного состояния и высушить при 100°
или более высокой температуре для удаления гигроскопической
влаги. Во многих случаях можно успешно использовать кристаллогидраты, если они
не гигроскопичны на воздухе и заметно не подвергаются выветриванию. Кристаллы
должны быть достаточно большими, чтобы можно было выбрать из них прозрачные и
невыветрившиеся. Такие воздушно-сухие кристаллы могут содержать несколько
десятых долей процента адсорбированной влаги, но это редко имеет значение при
определении следов. Слишком точное взвешивание стандартного вещества является
неоправданной тратой времени. Например, для приготовления 1 л
0,1%-ного раствора шестнвалентного
хрома следует взять 3,73 г. К.оСг04,
а не 3,734 г.
Отбор проб
При
колориметрическом определении следов обычно берут относительно большую пробу, и
отбор средней пробы, как правило, не представляет особых трудностей. Нужно
соблюдать лишь обычные меры предосторожности. Следует обращать внимание на то,
чтобы в анализируемый образец не попадали сопутствующие посторонние вещества.
Например, загрязнение пробы почвой или пылью может привести к существенным
ошибкам при определении следов веществ в растениях.
Приготовление биологических проб для определения следов металлов
За
очень редкими исключениями, органическую ткань необходимо разрушить, прежде чем
приступить к определению в ней следов элементов. Это можно сделать двумя
способами – сухим сжиганием или мокрым окислением. По первому методу образец
прокаливают в муфельной печи обычно при температуре 500–550°.
Этот метод имеет ряд преимуществ: выполнение его просто,
исключено попадание в образец следов посторонних элементов с окислителями,
применяемыми в мокром методе. Однако полное сожжение углерода не всегда
происходит быстро и легко; особенно доставляют трудности вещества животного
происхождения, и может иметь место потеря микроэлементов. Определенные элементы
могут улетучиваться. Существует также опасность, что следы определяемых
элементов прореагируют с материалом чашки. При озоленин образцов с низким
содержанием минеральных веществ в кварцевых или фарфоровых чашках заметная
часть определяемого компонента удерживается на поверхности чашки вследствие образования
силиката, который не всегда полностью удается разложить кислотами. Этот эффект
проявляется заметнее при пользовании старыми чашками с шероховатой
поверхностью. Снижению таких потерь способствует добавление инертных веществ
для уменьшения поверхности контакта. Если применить платиновую чашку, то легко
окисляемые металлы, такие, как свинец или медь, могут сплавляться со стенкой и
при промывке кислотой их невозможно удалить полностью. Нерастворимые вещества могут
прочно захватывать тяжелые металлы. Наконец, при сухом сжигании имеется
некоторая опасность загрязнения проб атмосферной пылью или веществами,
возгоняющимися с внутренней поверхности печи.
Удержание железа, алюминия и марганца двуокисью кремния при
сжигании образцов сена и соломы а)
В каждом случае указанным ниже способом сжигали образец
весом 5 г; золу обрабатывали разбавленной соляной кислотой, раствор
выпаривали досуха и остаток экстрагировали разбавленной соляной кислотой;
железо, алюминий и марганец определяли в промытом остатке после обработки
плавиковой кислотой.
Из-за
этих трудностей, или, иначе, неопределенностей, возникающих при сухом озолении
образцов, многие авторы предпочитают использовать целиком или частично мокрый
способ окисления для разрушения органического вещества. В качестве окислителей
почти всегда применяют кислоты, однако нет единодушного мнения относительно
наилучшего способа мокрого окисления. На самом деле различные материалы могут
требовать неодинаковой обработки. Углеводы разлагаются легко, белки и жиры
труднее. В методах мокрого окисления часто применяется смесь азотной и серной
кислот. Часто она дает удовлетворительные результаты, однако при обработке
животных тканей может потребоваться длительное кипячение, а некоторые образцы
никогда не образуют бесцветный раствор. Хлорная кислота в смеси с серной или
азотной кислотой более эффективна, чем смесь серной и азотной кислот, однако
даже она оказывается неудовлетворительной для некоторых стойких к окислению
веществ. Более того, многие исследователи воздерживаются от применения хлорной
кислоты как вещества потенциально опасного. Во всяком случае, применяя хлорную
кислоту для окисления, необходимо соблюдать осторожность, но по возможности
лучше обойтись без нее.
Разработан
метод, который применим к широкому ряду материалов биологического
происхождения. По существу он представляет окисление концентрированной или
дымящей азотной кислотой при относительно высокой температуре. Для
предотвращения возгорания материала в начальной стадии выпаривания добавляют
небольшое количество серной кислоты. Азотная кислота – единственная,
затрачиваемая в большом количестве, и, если необходимо, ее можно перегнать для
удаления примесей металлов. Считают, что этот метод особенно эффективен для
азотсодержащих тканей животного происхождения. Как полагают, он пригоден для
определения таких металлов, как медь, железо, цинк, марганец и алюминий, и в
частности, как было показано, дает удовлетворительные результаты при
определении свинца и кобальта.
|