Реферат: Активация алкенов и алкинов
Реферат: Активация алкенов и алкинов
АКТИВАЦИЯ АЛКЕНОВ И
АЛКИНОВ
1. π-Комплексы
алкенов и алкинов
Превращения олефинов и
ацетиленов относятся к важнейшим реакциям основного органического синтеза. Как
правило, эти превращения включают активацию π-лигандов в π-комплексах
переходных металлов. Фактически, в рамках тех понятий и терминов, которые мы
обсуждаем, π-комплексы являются первичными комплексами.
π-Комплексы
моноолефинов, сопряженных и несопряженных диолефинов и ацетиленов известны
почти для всех переходных металлов. При этом встречаются моно-, би- и
полиядерные соединения. Известны также соединения, содержащие 2, 3 и даже 4
молекулы олефина на атом металла, например, AcacRh(C2H4)2, Ni(C2H4)3 и Ir(C2H4)4Cl.
В ацетиленовых π-комплексах
состава Mm(C2R2)n соотношение m/n меняется весьма широко. Например,
известны комплексы состава Co4(CO)10(C2H2), Pt(C2R2)2, W(CO) (C2R2)3.
Координация всех π-лигандов
сопровождается более или менее значительными изменениями их физических
характеристик – понижается частота валентных колебаний кратных связей,
увеличивается длина связей С-С, изменяются величины валентных углов.
Природа химической связи в
π-комплексах переходных металлов имеет много общего. Рассмотрим основные
положения на примере π-комплексов олефинов и ацетиленов.
Молекула этилена по
величине потенциала ионизации не отличается от молекулы аммиака (IC2H4 = 10.5 эв). Донорные свойства ацетилена выражены
несколько слабее (IC2H2 = 11.4 эв). В ацетилене, однако, нижние вакантные МО
лежат ниже, чем у этилена, поэтому молекула ацетилена характеризуется более
выраженными акцепторными свойствами.
Представления о природе
связи в π-комплексах базируются на идеях Дьюара, Чатта и Данкансона,
развитых более полувека назад. Модель Дьюара-Чатта-Данкансона, несмотря на ее
упрощенность, может быть положена в основу концепции активации π-лигпндов
в комплексах переходных металлов.
Рассмотрим, например,
соль Цейзе KPtCl3(C2H4). Молекула этилена занимает координационное место в
плоско-квадратном комплексе PtCl42-, вытесняя ион хлора:
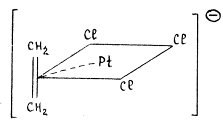
Гибридная орбиталь Pt(II) (например, dsp2-орбиталь)
перекрывается с π-МО этилена, образуя трехцентровую двухэлектронную связь
(донорно-акцепторная компонента связи). Гибридные заполненные 6dp-орбитали платины взаимодействуют с
разрыхляющей π*-МО олефина, образуя вторую трехцентровую двухэлектронную
связь (дативная компонента связи). В общем виде эти ситуация представлена на
диаграмме:
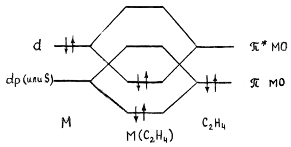
Обе трехцентровые связи
вносят вклад в повышение прочности связи M-C2H4. Однако эти компоненты по разному влияют на свойства
координированного π-лиганда.
В частности, образование
дативной связи увеличивает заселенность разрыхляющей орбитали π-лиганда,
что приводит к сильной дестабилизации координированной молекулы. Соотношение
двух компонент связи металл-лиганд будет зависеть от свойств центрального
атома, других лигандов и заместителей в молекуле π-лиганда. По степени
изменения свойств π-лиганда все π-комплексы делятся на две группы.
I тип – π-комплексы, в которых
изменение π-лиганда носит характер слабого возмущения.
II тип – π-комплексы, в которых
сильно меняются порядок связи С-С и валентные углы.
Комплексы I типа обычно образуются металлами в
высоких степенях окисления, связанных с электроотрицательными лигандами.
Комплексы II типа образуют металлы в низких
степенях окисления, связанные с мягкими основаниями. Так, например, соль Цейзе
– K[PtCl3(C2H4)] – является примером комплекса I типа, а соединение (Ph3P)2Pt(C2H4) – примером комплекса II типа (см. данные таблицы).
| Комплекс |
Тип |
ΔνС=С,
см-1
|
LC=C, Ǻ
|
Положение
С=С |
|
K[PtCl3(C2H4)]
(Ph3P)2Pt(C2H4)
|
I
II
|
118
> 200
|
1.37
1.43
|
Перпендикулярно
пл. PtCl3
В
плоскости Pt(PPh3)2
|
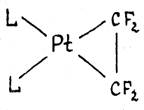
В комплексах c сильными электроотрицательными π-лигандами длины связей
С-С приближаются к длине простой связи С-С в алканах. Например, в комплексе L2Pt(C2F4) длина связи С-С составляет 1.54 Ǻ, т.е. двойная
связь по существу превращается в одинарную. Значения химсдвигов 19F и констант спин-спинового взаимодействия в спектре ЯМР
комплекса также хорошо объясняются “циклопропановой” структурой.
Т.о., взаимодействие
сильного донора L2Pt (0) с сильным акцептором C2F4 приводит к существенному переносу d-электронов на π-лиганд и образованию металлоцикла.
Происходит окислительное присоединение π-лиганда к Pt(0) с образованием Pt(II) и аниона C2F42-.
Особенно отчетливо
различие двух групп π-комплексов можно проследить в случае ацетиленовых
лигандов (см. таблицу).
| Комплекс |
Тип |
ΔνС=С,
см-1
|
LC=C, Ǻ
|
Положение
С=С |
|
(Bu2C2PtCl2L*
(Ph2C2)Pt(PPh3)2
|
I
II
|
200
490
|
1.27
1.36
|
Перпендикулярно
пл. PtCl3
В
плоскости Pt(PPh3)2
|
2. ПУТИ АКТИВАЦИИ
АЛКЕНОВ
Наиболее характерной
реакцией π-комплексов олефинов, относящихся к I типу, является реакция с нуклеофильными реагентами.
Нуклеофильный реагент
часто атакует π-комплекс со стороны, противоположной металлу
(транс-присоединение), образуя σ-металлоорганическое соединение:
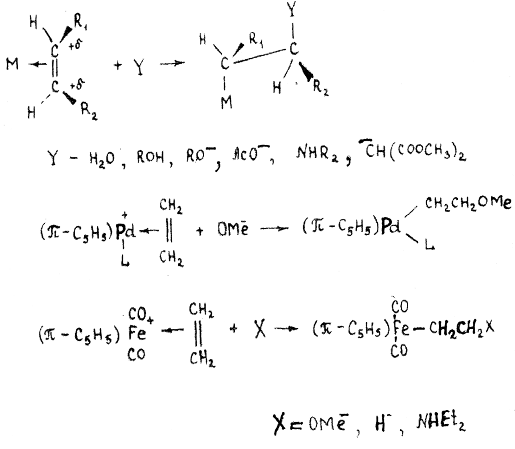
В качестве нуклеофилов
могут выступать анионные карбонилметаллаты, образуя σ-металлоорганические
соединения с мостиковым этиленом:
(π-C5H5)(CO)3M(π-C2H4)
+ Re(CO)5- → (π-C5H5)(CO)3MCH2CH2Re(CO)5
M = Mo, W
Поляризация молекулы
олефина в π-комплексе может приводить к смещению электронов в группах,
соседних с С=С – связью, что проявляется, например, в образовании π-аллильных
комплексов:
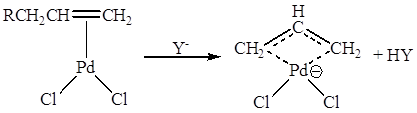
В π-комплексах II типа характер реакционной
способности кратной связи меняется. π-Лиганд становится способным
взаимодействовать с электрофильными реагентами, например в соответствии со схемой:
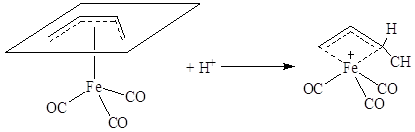
Более характерными для π-комплексов
II типа являются реакции, связанные с
общим разрыхлением всей молекулы из-за переноса электронов на олефин. К таким
реакциям следует отнести реакции циклообразования, внедрения по связи
металл-металл, окислительного присоединения или замещения по связи =С-Х (где X = H, Cl, F).
Например:
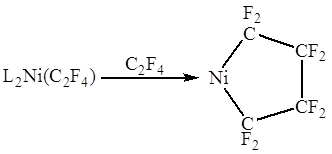
Или
(CO)4Co-Co(CO)4
+ CF2=CF2 → (CO)4Co- CF2CF2-Co(CO)4
Иногда активация олефина
в π-комплексах II типа настолько
лабилизирует связи в π-лиганде, что становится возможным разрыв связей С-Н
при двойной связи и в соседней с двойной связью метильной группе, приводящий к
продуктам окислительного присоединения:
Os3(CO)12
+ C2H4 → H2Os3(C=CH2)(CO)9
+ 3CO
3. ПУТИ АКТИВАЦИИ АЛКИНОВ
В ацетиленовых комплексах
I типа (Ag(I), Cu(I), Hg(II), Pt(II), Pd(II), Ru(III) и др.) повышение электрофильности
тройной связи приводит к облегчению взаимодействия с нуклеофильной частицей из
раствора (транс-присоединение) или нуклеофилом, координированным
металлом (цис-внедрение):
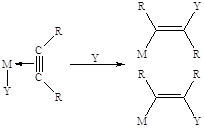
Основные реакции π-комплексов
I типа довольно удачно промоделированы
на комплексах Pt(II):
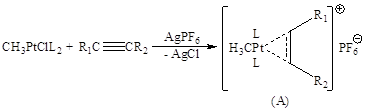
Образующийся катионный π-комплекс
способен превращаться по нескольким направлениям:
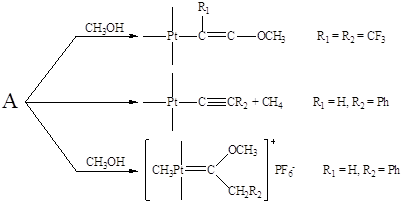
В π-комплексах II типа в первую
очередь сильно разрыхляется тройная связь углерод-углерод и связь C-X при тройной связи. Так, активация связи С-Н в π-комплексах
Ni(0), Pt(0), Os(0)
или Rh(I) приводит к окислительному присоединению с образованием
этинилгидридного комплекса металла:
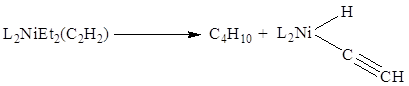
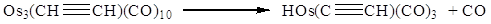
Вторая характерная
реакция для π-комплексов II
типа – это реакция циклообразования, причем в состав получающегося металлоцикла
входят уже две молекулы алкина:
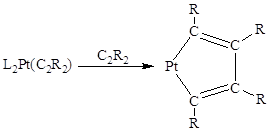
Металлоциклопентадиены из
алкинов получены в реакциях комплексов Pt(0), Pd(0),
Co(I), Fe(0),
Rh(I), Ir(I) и Ti(II).
Активация полярных
молекул
Полярные молекулы НХ, где
Х – ОН, OR, Hal, CN, NH2, NR2, NHR, SR и др. активируются по механизму, близкому к механизму
активации апротонными кислотами. Образование донорно-акцепторной связи между
донорным атомом полярной молекулы и комплексом переходного металла, имеющим
вакантные орбитали, приводит к ослаблению связи Н – Х. Ослабление связи H-X при координации этих молекул подтверждается, как правило,
данными ИК-спектров координированных молекул. При этом в образующемся комплексе
происходит смещение σ-пары электронов донорных атомов O, N или S к
иону металла, обладающему акцепторными свойствами. Молекула лиганда
поляризуется, что приводит к ее ионизации и облегчает диссоциацию (в полярных
растворителях):
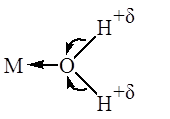
Смещение электронов и
ослабление связи Х-Н при координации подтверждается данными ИК-спектров
координированных молекул. Например, координация RNH2 в комплексах с PtCl2 приводит к понижению νN-H (на 80-100 см-1).
В результате повышается
способность связи Н-Х к гетеролитической диссоциации с передачей протона на
другой субстрат или его окислительному присоединению к переходному металлу (в
зависимости от степени окисления металла и состояния его внутренней
координационной сферы).
HX + MLn
—→ H--X→MLn-1 + L
Даже такие слабые
кислоты, как молекула аммиака или аминов, легко депротонируются в водных или
неводных средах в координационной сфере переходных металлов:
TiCl4
+ 6NH3 → TiCl(NH2)3 + 3 NH4Cl
Или
Pt(NH3)X5-
+ H2O → Pt(NH3)X52- + H3O+
В результате в комплексе
металла появляется фрагмент молекулы HX (например, NH2), реакционная способность которого,
конечно, ниже, чем свободного иона NH2-, но
концентрация которого на много порядков выше, чем в отсутствие
комплексообразователя.
Константа диссоциации
ацетонитрила HCN, например, составляет 10-10.
Образование комплексов с металлами позволяет существенно повысить концентрацию
группы M-CN.
Так, при взаимодействии HCN с полиядерными комплексами меди(I)
CumCln(n-m)- + HCN → ClnCum-1(CuCN) + HCl
концентрация CuCN в растворе может достигать 15% вес. Координированный
анион CN может далее участвовать в различных
реакциях:
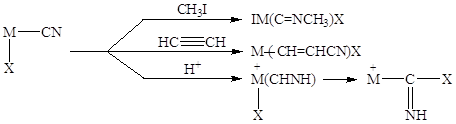
Для катализа особенно
важны две последние реакции. Последняя реакция приводит к образованию
изосинильной кислоты, способной внедряться по связи M-X.
Рекомендуемая
литература
1.
Г. Хенрици-Оливэ,
С. Оливэ. Химия каталитического гидрирования СО. Москва, Мир, 1987 г.
2.
Ф. Басоло, Р.
Джонсон. Химия координационных соединений. Москва, Мир, 1966.
3.
Под ред. Г.
Цейсса. Химия металлоорганических соединений. Москва, Мир, 1964.
4.
Э. Фишер, Г.
Вернер. π-Комплексы металлов. Москва, Мир, 1968.
|